
Differential Gene Expression#
In single cell, differential expresison can have multiple functionalities such as of identifying marker genes for cell populations, as well as differentially regulated genes across conditions (healthy vs control). We will also exercise on how to account the batch information in your test.
Differential expression is performed with the function rank_genes_group. The default method to compute differential expression is the t-test_overestim_var. Other implemented methods are: logreg, t-test and wilcoxon.
By default, the .raw attribute of AnnData is used in case it has been initialized, it can be changed by setting use_raw=False.
The clustering with resolution 0.6 seems to give a reasonable number of clusters, so we will use that clustering for all DE tests.
For Differential Expression we need all genes and
#@title Load Filtered, Integrated clustered data
adata = sc.read_h5ad('Objects/sc_QCNFSDM_scCorrected__clustered_covid.h5ad')
adata
AnnData object with n_obs × n_vars = 9000 × 2000
obs: 'type', 'sample', 'batch', 'leiden_1.0', 'leiden_0.6', 'leiden_0.4', 'leiden_1.4', 'louvain_1.0', 'louvain_0.6', 'louvain_0.4', 'louvain_1.4', 'kmeans5', 'kmeans10', 'kmeans15'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'dendrogram_leiden_0.6', 'dendrogram_louvain_0.4', 'dendrogram_louvain_0.6', 'dendrogram_louvain_1.0', 'hvg', 'kmeans10_colors', 'kmeans15_colors', 'kmeans5_colors', 'leiden', 'leiden_0.4_colors', 'leiden_0.6_colors', 'leiden_1.0_colors', 'leiden_1.4_colors', 'log1p', 'louvain', 'louvain_0.4_colors', 'louvain_0.6_colors', 'louvain_1.0_colors', 'louvain_1.4_colors', 'neighbors', 'pca', 'sample_colors', 'tsne', 'type_colors', 'umap'
obsm: 'Scanorama', 'X_pca', 'X_tsne', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'connectivities', 'distances'
#@title load Raw Data
adata.raw = sc.read_h5ad('/content/drive/MyDrive/scRNA_using_Python/Objects/adata_raw_covid.h5ad')
adata.raw
<anndata._core.raw.Raw at 0x7fa992628e80>
print(adata.raw.X[:10,:10])
print(adata.X.shape)
print(adata.raw.X.shape)
(9000, 2000)
(9000, 33538)
adata_raw
AnnData object with n_obs × n_vars = 9000 × 21830
obs: 'type', 'sample', 'batch', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'sample_colors', 'type_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'logcounts'
obsp: 'distances', 'connectivities'
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch', 'leiden_1.0', 'leiden_0.6', 'leiden_0.4', 'leiden_1.4', 'louvain_1.0', 'louvain_0.6', 'louvain_0.4', 'louvain_1.4', 'kmeans5', 'kmeans10', 'kmeans15', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dendrogram_leiden_0.6', 'dendrogram_louvain_0.4', 'dendrogram_louvain_0.6', 'dendrogram_louvain_1.0', 'hvg', 'kmeans10_colors', 'kmeans15_colors', 'kmeans5_colors', 'leiden', 'leiden_0.4_colors', 'leiden_0.6_colors', 'leiden_1.0_colors', 'leiden_1.4_colors', 'log1p', 'louvain', 'louvain_0.4_colors', 'louvain_0.6_colors', 'louvain_1.0_colors', 'louvain_1.4_colors', 'neighbors', 'pca', 'sample_colors', 'tsne', 'type_colors', 'umap', 't-test', 't-test_ov', 'wilcoxon'
obsm: 'Scanorama', 'X_pca', 'X_tsne', 'X_umap'
obsp: 'connectivities', 'distances'
For DGE analysis we would like to run with all genes, but on normalized values, so we will have to revert back to the raw matrix and renormalize.
We can’t see Louvain_0.6 but it exist
adata = adata.raw.to_adata()
sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
sc.pp.log1p(adata)
normalizing by total count per cell
finished (0:00:00): normalized adata.X and added 'n_counts', counts per cell before normalization (adata.obs)
WARNING: adata.X seems to be already log-transformed.
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch', 'leiden_1.0', 'leiden_0.6', 'leiden_0.4', 'leiden_1.4', 'louvain_1.0', 'louvain_0.6', 'louvain_0.4', 'louvain_1.4', 'kmeans5', 'kmeans10', 'kmeans15', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dendrogram_leiden_0.6', 'dendrogram_louvain_0.4', 'dendrogram_louvain_0.6', 'dendrogram_louvain_1.0', 'hvg', 'kmeans10_colors', 'kmeans15_colors', 'kmeans5_colors', 'leiden', 'leiden_0.4_colors', 'leiden_0.6_colors', 'leiden_1.0_colors', 'leiden_1.4_colors', 'log1p', 'louvain', 'louvain_0.4_colors', 'louvain_0.6_colors', 'louvain_1.0_colors', 'louvain_1.4_colors', 'neighbors', 'pca', 'sample_colors', 'tsne', 'type_colors', 'umap'
obsm: 'Scanorama', 'X_pca', 'X_tsne', 'X_umap'
obsp: 'connectivities', 'distances'
sc.pl.umap(adata, color='louvain_0.6')

Tests#
T-test#
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='t-test', key_added = "t-test")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "t-test")
---------------------------------------------------------------------------
NameError Traceback (most recent call last)
<ipython-input-1-7595d372b507> in <cell line: 1>()
----> 1 sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='t-test', key_added = "t-test")
2 sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "t-test")
NameError: name 'sc' is not defined
# results are stored in the adata.uns["t-test"] slot
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch', 'leiden_1.0', 'leiden_0.6', 'leiden_0.4', 'leiden_1.4', 'louvain_1.0', 'louvain_0.6', 'louvain_0.4', 'louvain_1.4', 'kmeans5', 'kmeans10', 'kmeans15', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dendrogram_leiden_0.6', 'dendrogram_louvain_0.4', 'dendrogram_louvain_0.6', 'dendrogram_louvain_1.0', 'hvg', 'kmeans10_colors', 'kmeans15_colors', 'kmeans5_colors', 'leiden', 'leiden_0.4_colors', 'leiden_0.6_colors', 'leiden_1.0_colors', 'leiden_1.4_colors', 'log1p', 'louvain', 'louvain_0.4_colors', 'louvain_0.6_colors', 'louvain_1.0_colors', 'louvain_1.4_colors', 'neighbors', 'pca', 'sample_colors', 'tsne', 'type_colors', 'umap', 't-test'
obsm: 'Scanorama', 'X_pca', 'X_tsne', 'X_umap'
obsp: 'connectivities', 'distances'
T-test overestimated_variance#
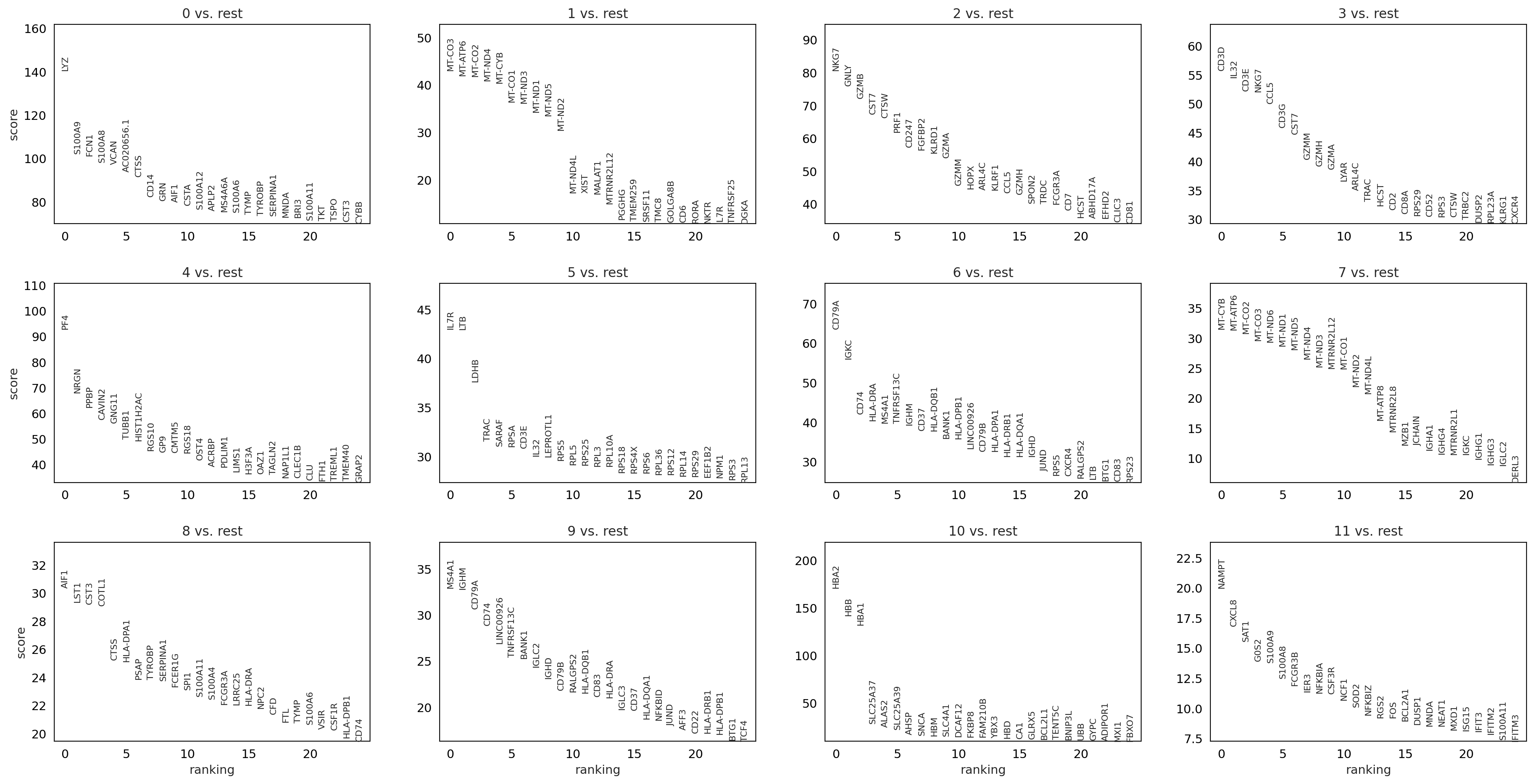
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='t-test_overestim_var', key_added = "t-test_ov")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "t-test_ov")
ranking genes
finished: added to `.uns['t-test_ov']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:01)
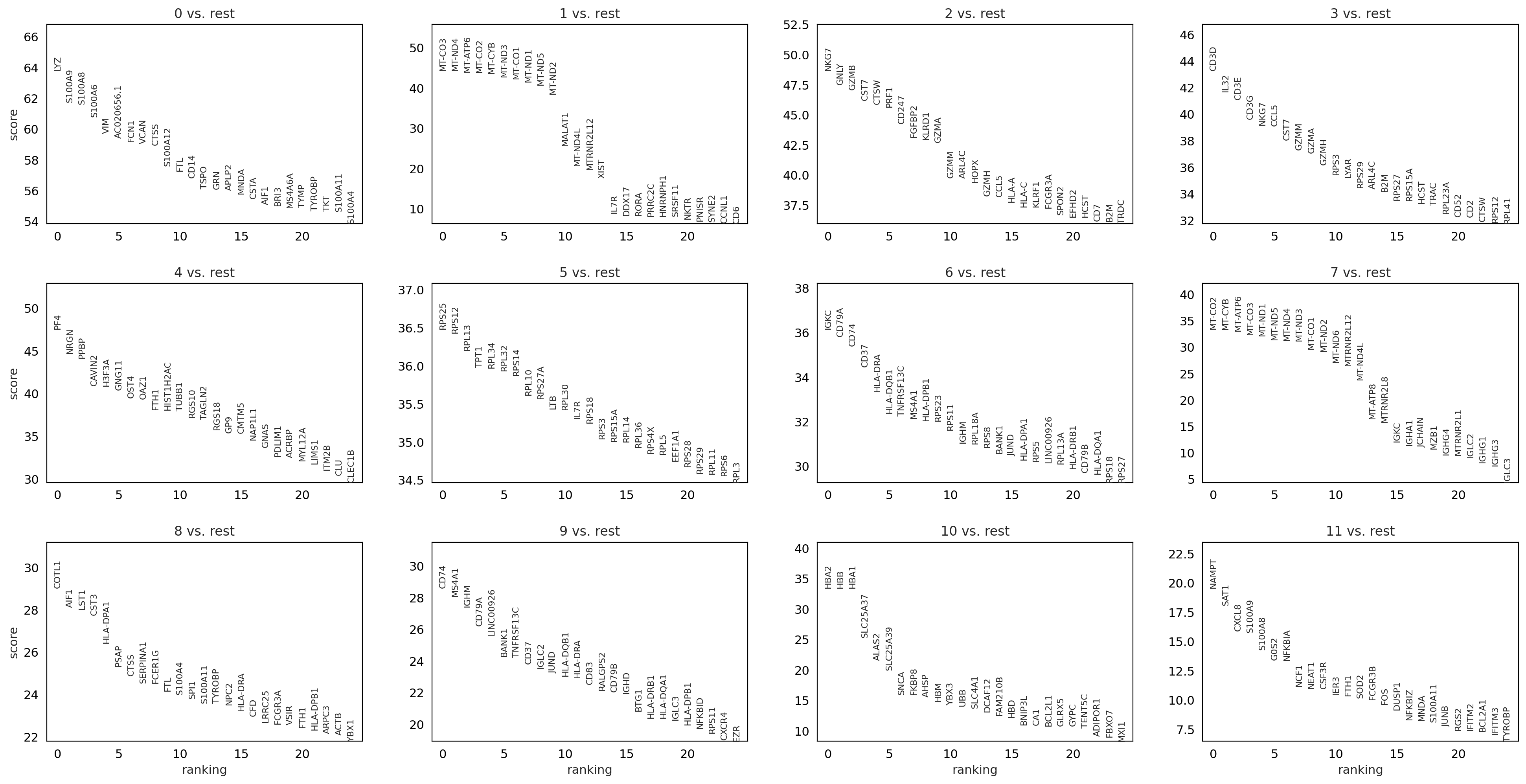
Wilcoxon rank-sum#
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key="wilcoxon")
ranking genes
finished: added to `.uns['wilcoxon']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:33)
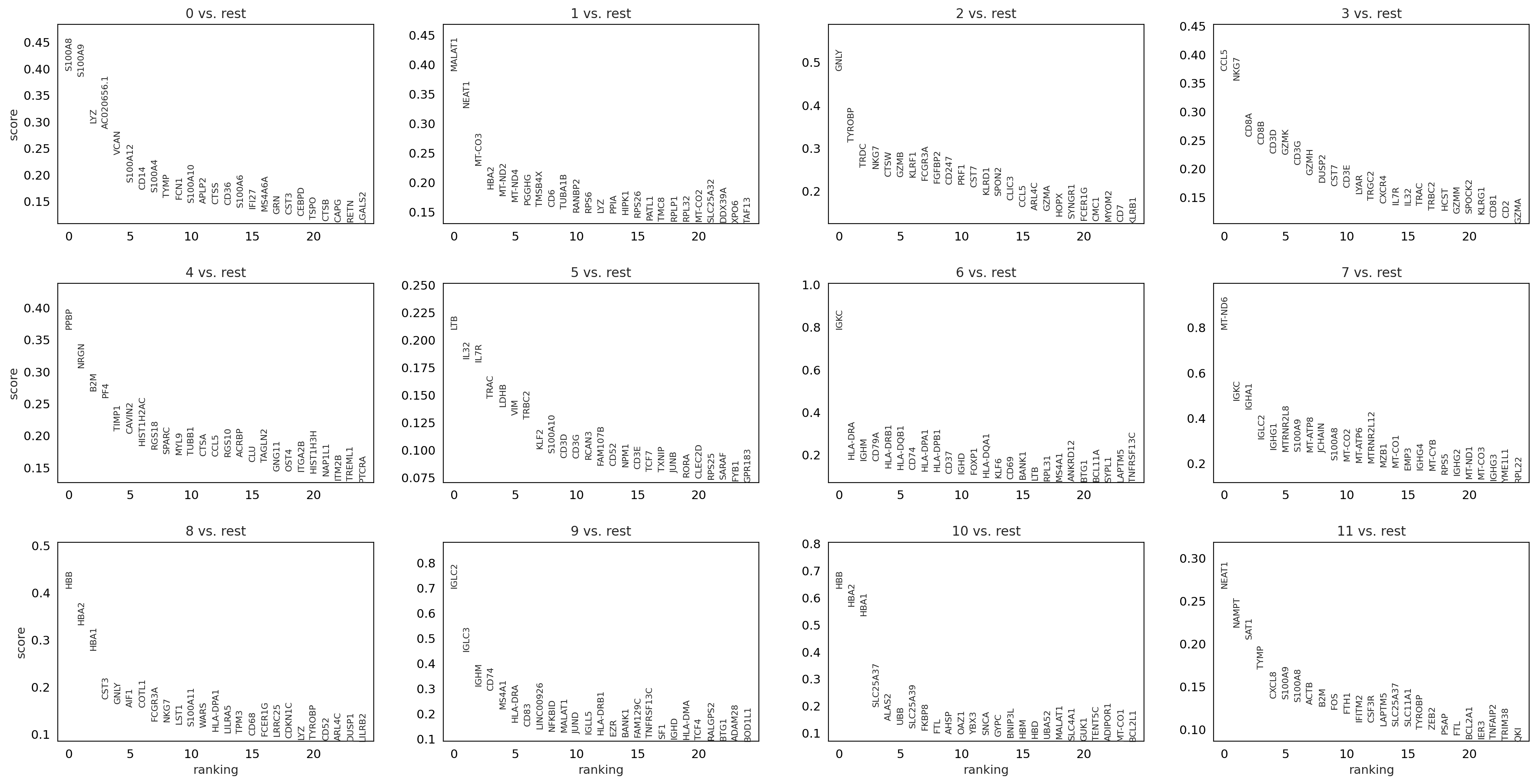
logistic regression (logres)#
sc.tl.rank_genes_groups(adata, 'louvain_0.6', method='logreg',key_added = "logreg")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False, key = "logreg")
ranking genes
finished: added to `.uns['logreg']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
(0:00:58)
Compare Genes#
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch', 'leiden_1.0', 'leiden_0.6', 'leiden_0.4', 'leiden_1.4', 'louvain_1.0', 'louvain_0.6', 'louvain_0.4', 'louvain_1.4', 'kmeans5', 'kmeans10', 'kmeans15', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dendrogram_leiden_0.6', 'dendrogram_louvain_0.4', 'dendrogram_louvain_0.6', 'dendrogram_louvain_1.0', 'hvg', 'kmeans10_colors', 'kmeans15_colors', 'kmeans5_colors', 'leiden', 'leiden_0.4_colors', 'leiden_0.6_colors', 'leiden_1.0_colors', 'leiden_1.4_colors', 'log1p', 'louvain', 'louvain_0.4_colors', 'louvain_0.6_colors', 'louvain_1.0_colors', 'louvain_1.4_colors', 'neighbors', 'pca', 'sample_colors', 'tsne', 'type_colors', 'umap', 't-test', 'logreg', 'wilcoxon', 't-test_ov'
obsm: 'Scanorama', 'X_pca', 'X_tsne', 'X_umap'
obsp: 'connectivities', 'distances'
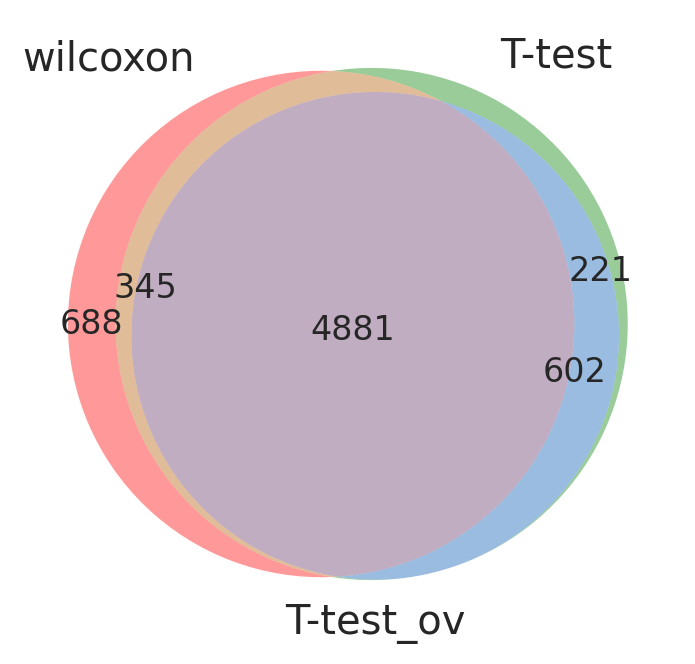
Take all significant DE genes for cluster0 with each test and compare the overlap
#compare cluster1 genes, only stores top 100 by default
wc = sc.get.rank_genes_groups_df(adata, group='0', key= 'wilcoxon', pval_cutoff=0.01, log2fc_min=0)['names']
tt = sc.get.rank_genes_groups_df(adata, group='0', key='t-test', pval_cutoff=0.01, log2fc_min=0)['names']
tt_ov = sc.get.rank_genes_groups_df(adata, group='0', key='t-test_ov', pval_cutoff=0.01, log2fc_min=0)['names']
from matplotlib_venn import venn3
venn3([set(wc),set(tt),set(tt_ov)], ('wilcoxon','T-test','T-test_ov') )
plt.show()
#@title Heatmap of genes
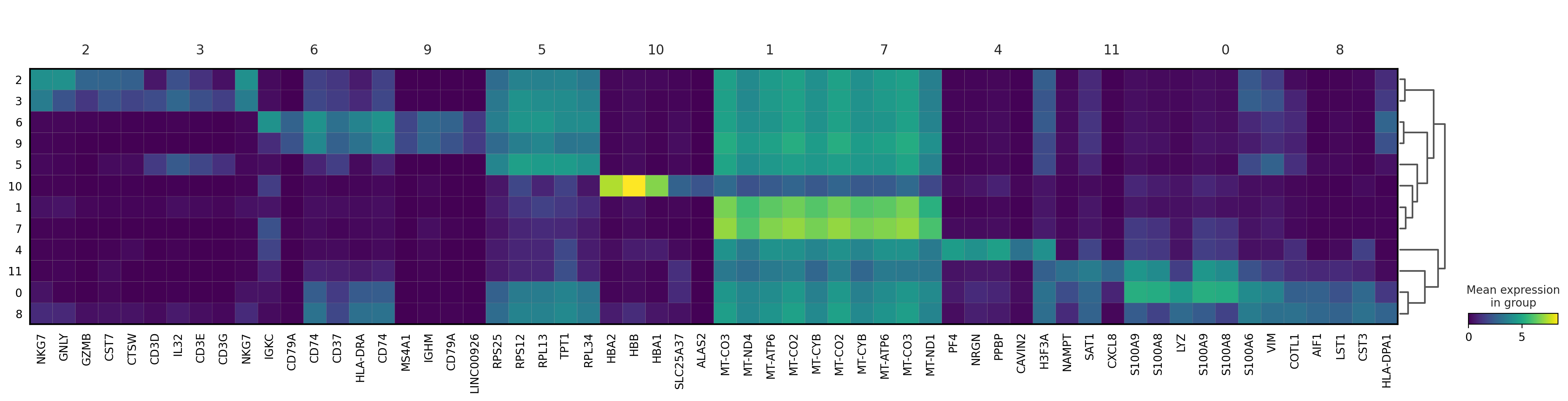
sc.pl.rank_genes_groups_heatmap(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6", show_gene_labels=True)
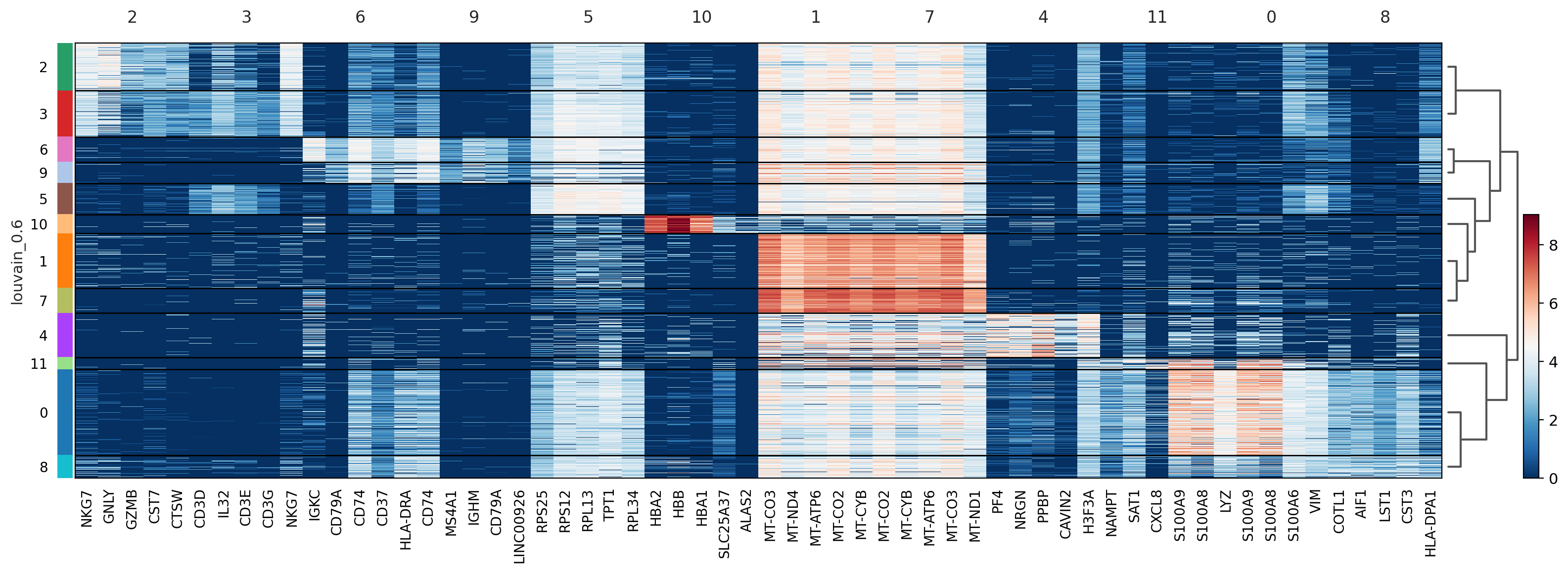
sc.pl.rank_genes_groups_dotplot(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6")
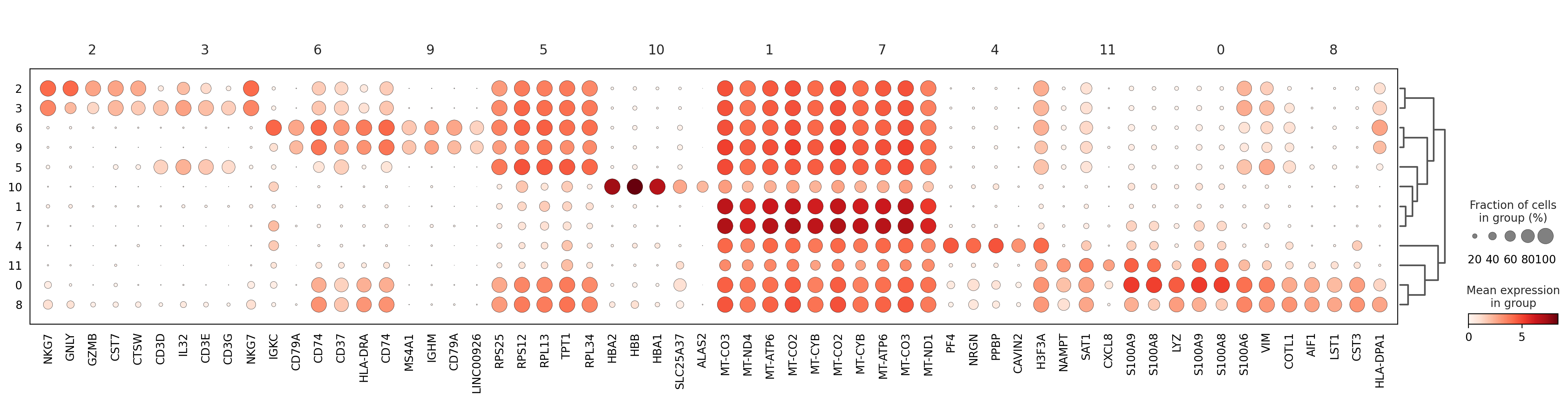
sc.pl.rank_genes_groups_stacked_violin(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6")
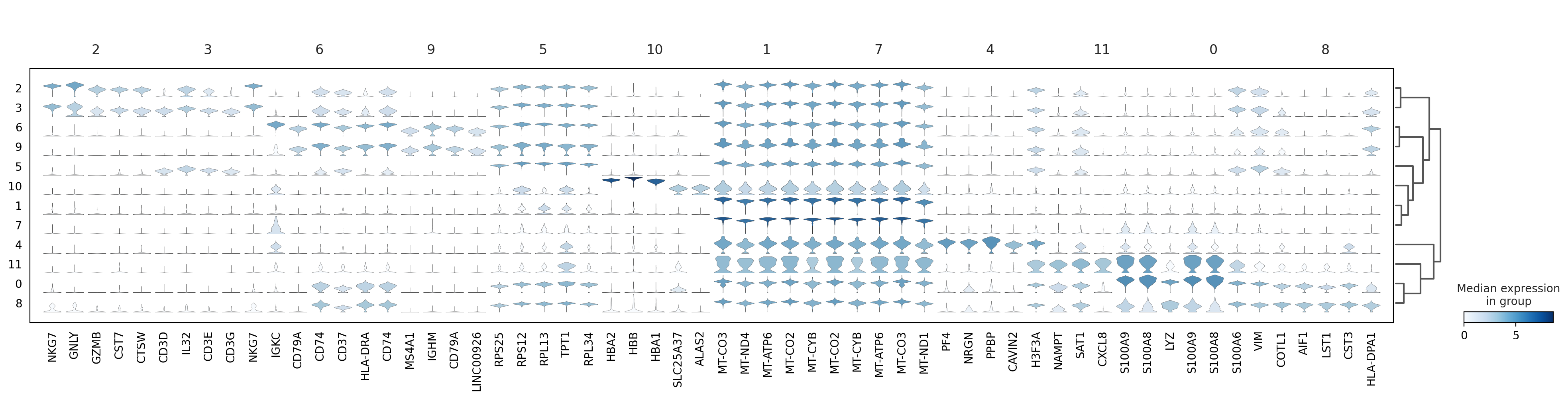
sc.pl.rank_genes_groups_matrixplot(adata, n_genes=5, key="wilcoxon", groupby="louvain_0.6")
Compare specific clusters#
#@title cluster1 vs cluster2
sc.tl.rank_genes_groups(adata, 'louvain_0.6', groups=['1'], reference='2', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['1'], n_genes=20)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:04)
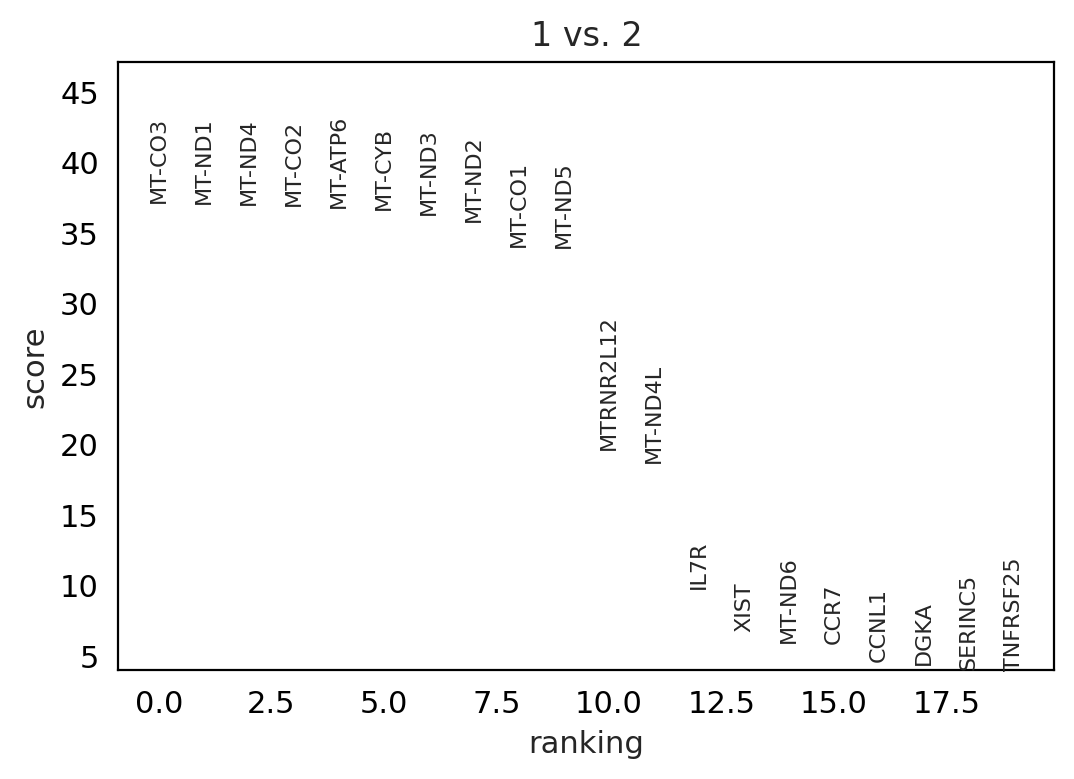
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch', 'leiden_1.0', 'leiden_0.6', 'leiden_0.4', 'leiden_1.4', 'louvain_1.0', 'louvain_0.6', 'louvain_0.4', 'louvain_1.4', 'kmeans5', 'kmeans10', 'kmeans15', 'n_counts'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'dendrogram_leiden_0.6', 'dendrogram_louvain_0.4', 'dendrogram_louvain_0.6', 'dendrogram_louvain_1.0', 'hvg', 'kmeans10_colors', 'kmeans15_colors', 'kmeans5_colors', 'leiden', 'leiden_0.4_colors', 'leiden_0.6_colors', 'leiden_1.0_colors', 'leiden_1.4_colors', 'log1p', 'louvain', 'louvain_0.4_colors', 'louvain_0.6_colors', 'louvain_1.0_colors', 'louvain_1.4_colors', 'neighbors', 'pca', 'sample_colors', 'tsne', 'type_colors', 'umap', 't-test', 'logreg', 'wilcoxon', 't-test_ov', 'rank_genes_groups'
obsm: 'Scanorama', 'X_pca', 'X_tsne', 'X_umap'
obsp: 'connectivities', 'distances'
sc.pl.rank_genes_groups_violin(adata, groups='1', n_genes=10)
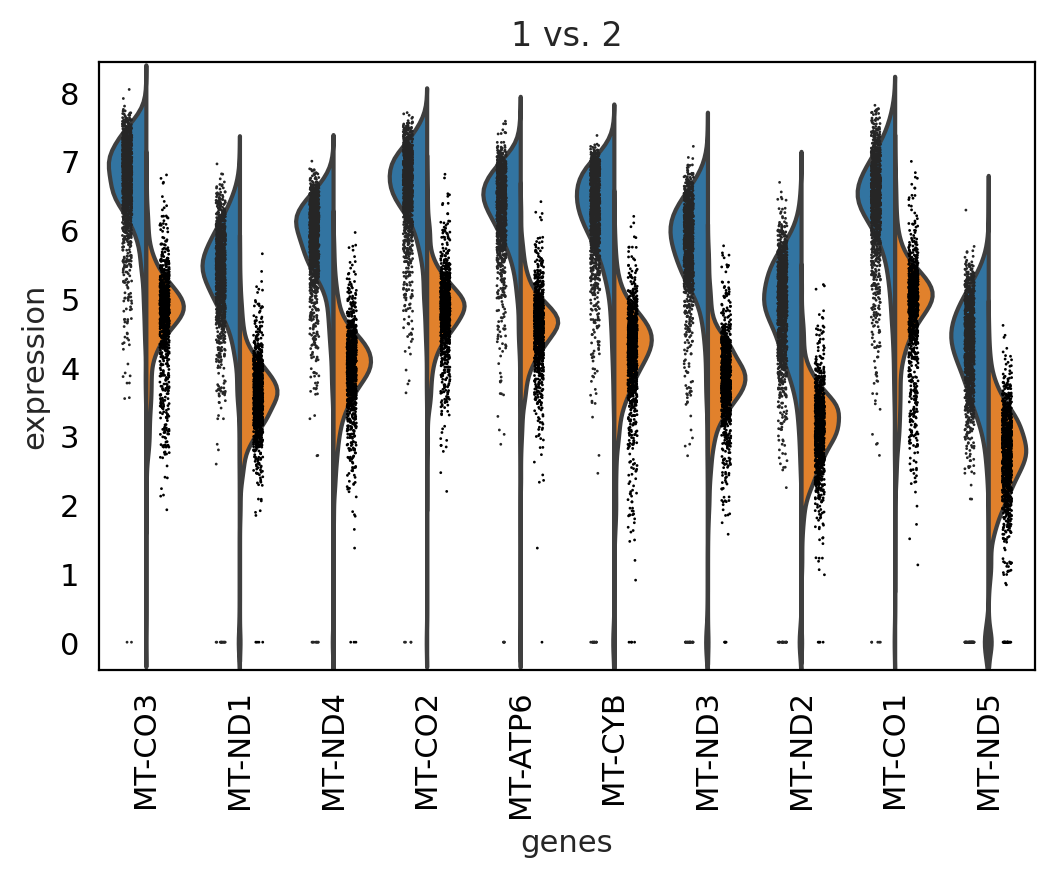
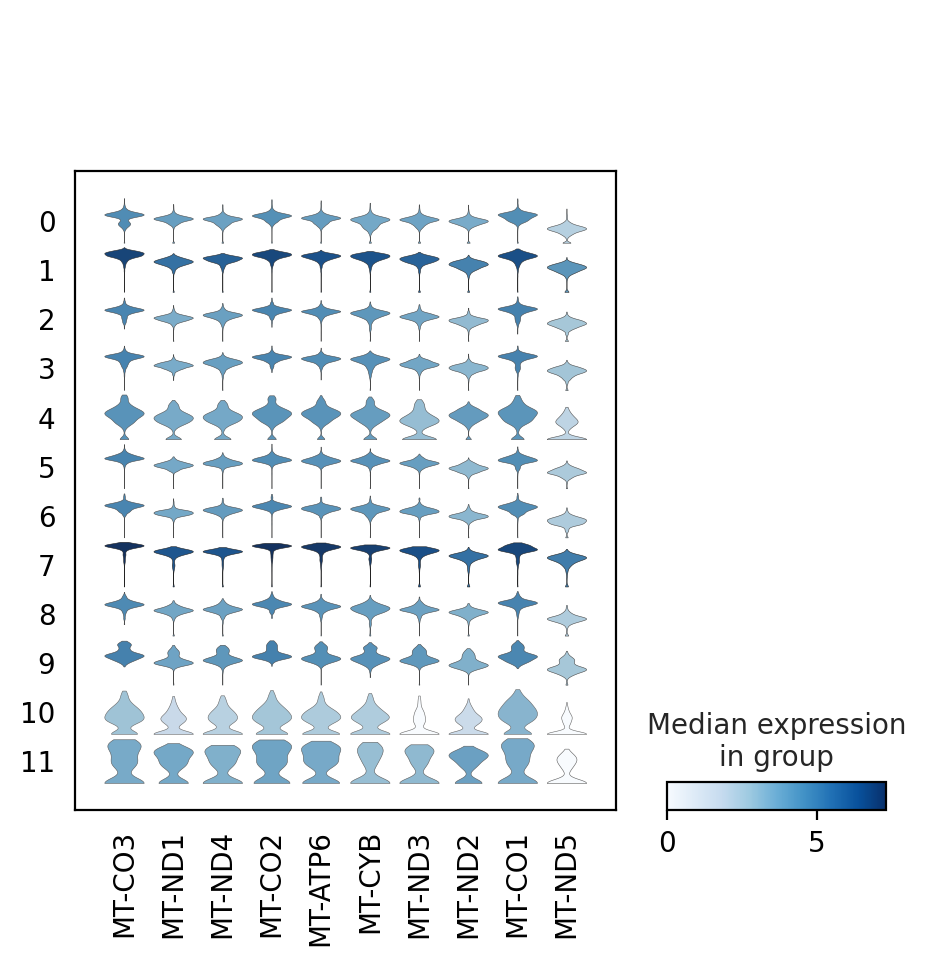
# plot the same genes as violins across all the datasets.
# convert numpy.recarray to list
mynames = [x[0] for x in adata.uns['rank_genes_groups']['names'][:10]]
sc.pl.stacked_violin(adata, mynames, groupby = 'louvain_0.6')
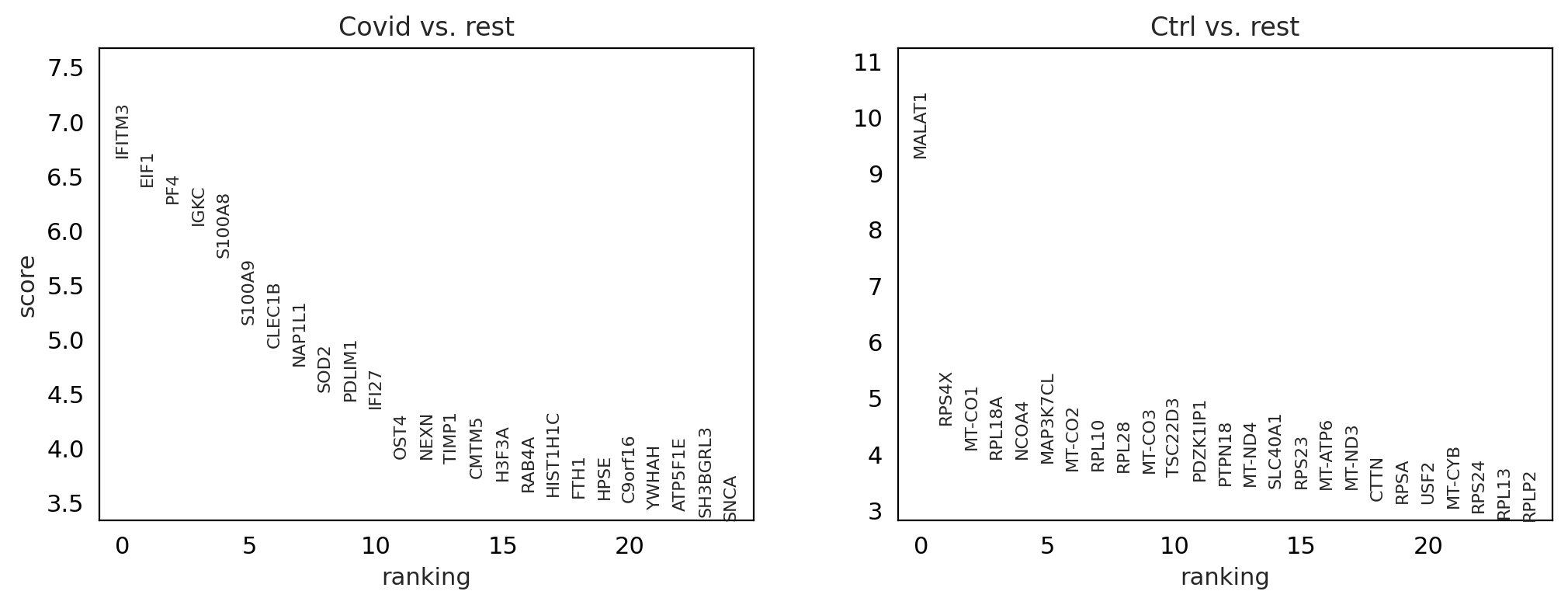
Differential expression across conditions#
The second way of computing differential expression is to answer which genes are differentially expressed within a cluster. For example, in our case we have libraries comming from patients and controls and we would like to know which genes are influenced the most in a particular cell type.
For this end, we will first subset our data for the desired cell cluster, then change the cell identities to the variable of comparison (which now in our case is the “type”, e.g. Covid/Ctrl).
cl1 = adata[adata.obs['louvain_0.6'] == '4',:]
cl1.obs['type'].value_counts()
Covid 858
Ctrl 54
Name: type, dtype: int64
sc.tl.rank_genes_groups(cl1, 'type', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(cl1, n_genes=25, sharey=False, key="wilcoxon")
ranking genes
finished: added to `.uns['wilcoxon']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:02)
sc.pl.rank_genes_groups_violin(cl1, n_genes=10, key="wilcoxon")
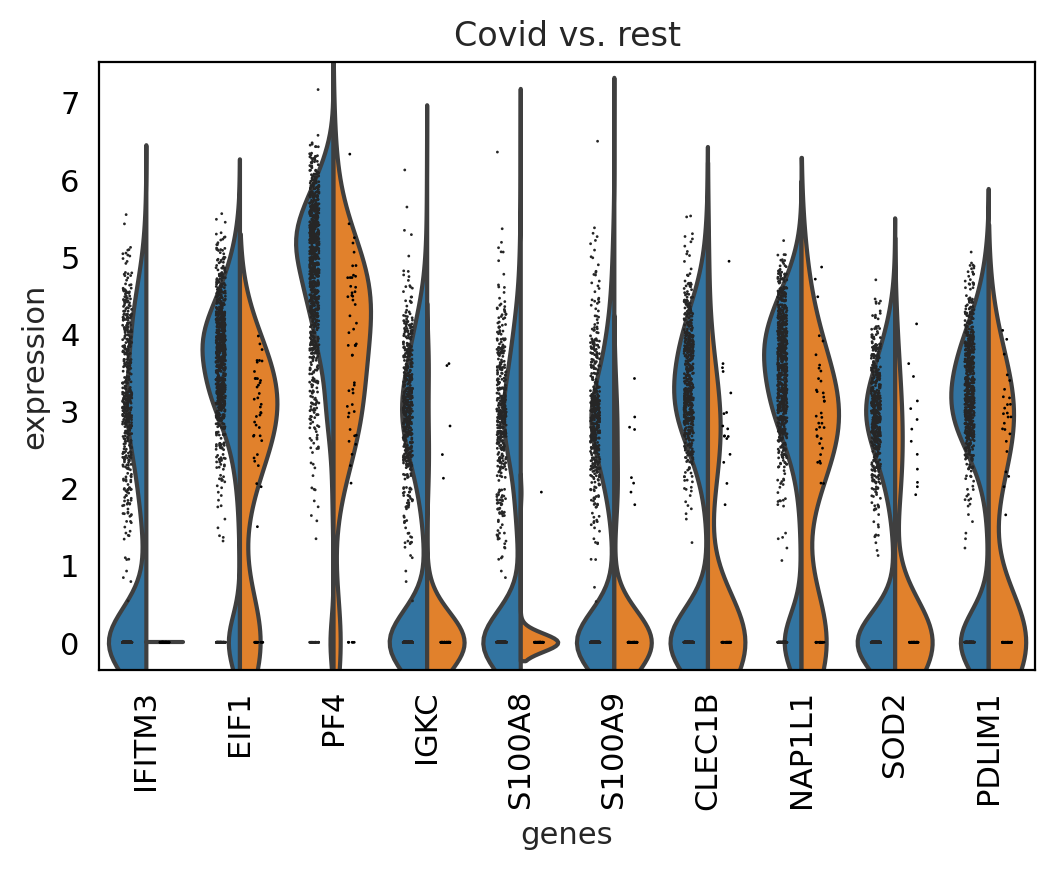
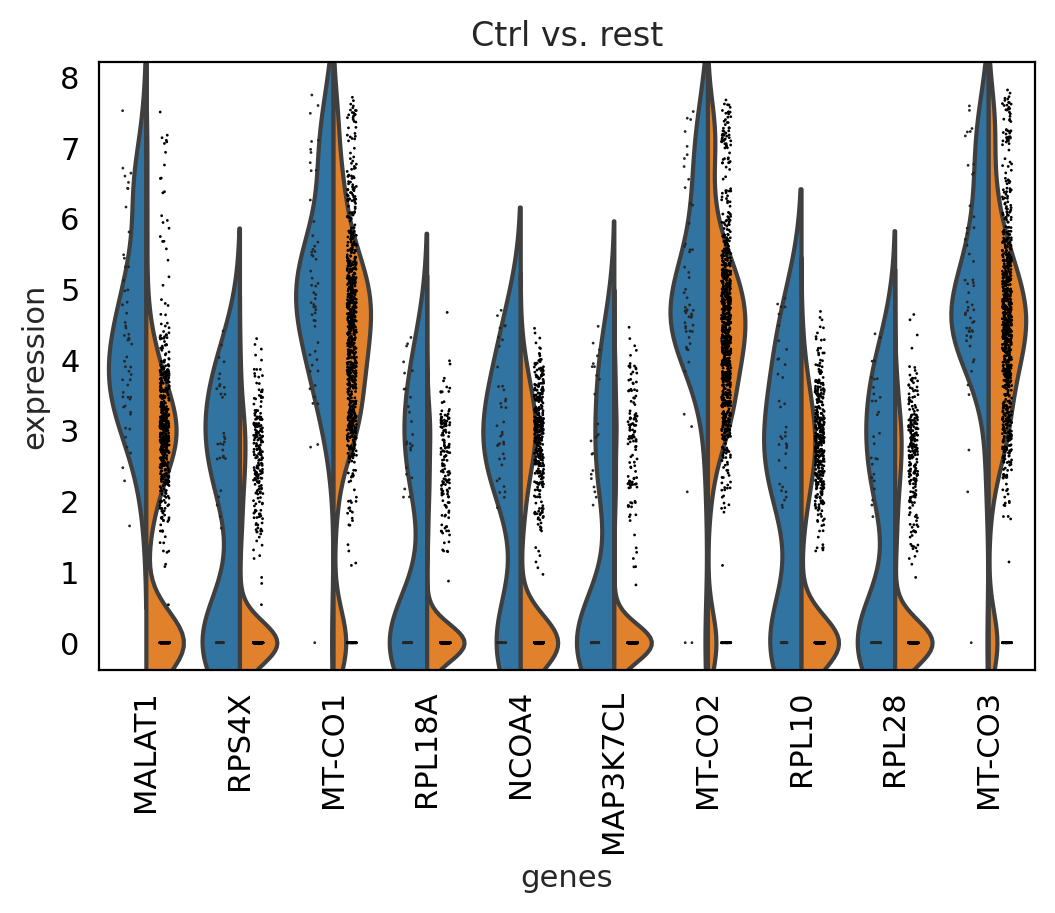
between cluster#
genes1 = sc.get.rank_genes_groups_df(cl1, group='Covid', key='wilcoxon')['names'][:5]
genes2 = sc.get.rank_genes_groups_df(cl1, group='Ctrl', key='wilcoxon')['names'][:5]
genes = genes1.tolist() + genes2.tolist()
df = sc.get.obs_df(adata, genes + ['louvain_0.6','type'], use_raw=False)
df2 = df.melt(id_vars=["louvain_0.6",'type'], value_vars=genes)
df.head(3)
| IFITM3 | EIF1 | PF4 | IGKC | S100A8 | MALAT1 | RPS4X | MT-CO1 | RPL18A | NCOA4 | louvain_0.6 | type | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | 0.000000 | 3.430040 | 0.832491 | 2.727161 | 0.832491 | 6.177206 | 3.019721 | 4.874463 | 3.781191 | 0.000000 | 9 | Covid |
| TACCCACAGCGGGTTA-1-0 | 1.381662 | 3.326245 | 1.174380 | 0.556972 | 5.466116 | 5.035670 | 3.118657 | 4.512954 | 3.403520 | 1.553268 | 0 | Covid |
| CCCAACTTCATATGGC-1-0 | 0.000000 | 3.404340 | 2.370070 | 0.473830 | 5.096470 | 5.208613 | 3.500291 | 5.054821 | 3.424281 | 1.534025 | 0 | Covid |
df2.head(3)
| louvain_0.6 | type | variable | value | |
|---|---|---|---|---|
| 0 | 9 | Covid | IFITM3 | 0.000000 |
| 1 | 0 | Covid | IFITM3 | 1.381662 |
| 2 | 0 | Covid | IFITM3 | 0.000000 |
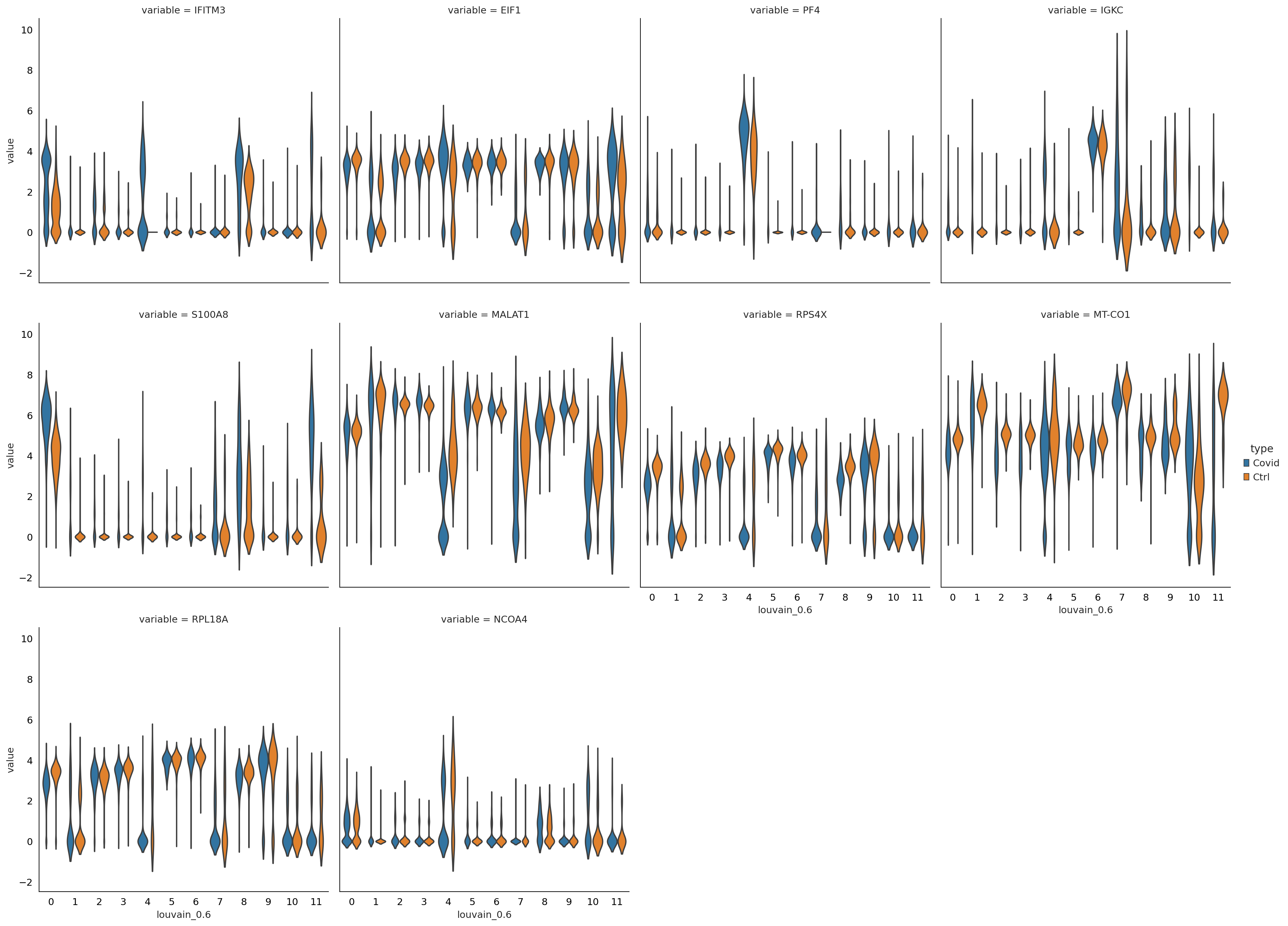
sns.catplot(x = "louvain_0.6", y = "value", hue = "type", kind = 'violin', col = "variable", data = df2, col_wrap=4, inner=None)
<seaborn.axisgrid.FacetGrid at 0x7fa9747897b0>
#@title remove sex genes and rerun differential expression
annot = sc.queries.biomart_annotations(
"hsapiens",
["ensembl_gene_id", "external_gene_name", "start_position", "end_position", "chromosome_name"],
).set_index("external_gene_name")
chrY_genes = adata.var_names.intersection(annot.index[annot.chromosome_name == "Y"])
chrX_genes = adata.var_names.intersection(annot.index[annot.chromosome_name == "X"])
sex_genes = chrY_genes.union(chrX_genes)
print(len(sex_genes))
all_genes = cl1.var.index.tolist()
print(len(all_genes))
keep_genes = [x for x in all_genes if x not in sex_genes]
print(len(keep_genes))
cl1 = cl1[:,keep_genes]
991
33538
32547
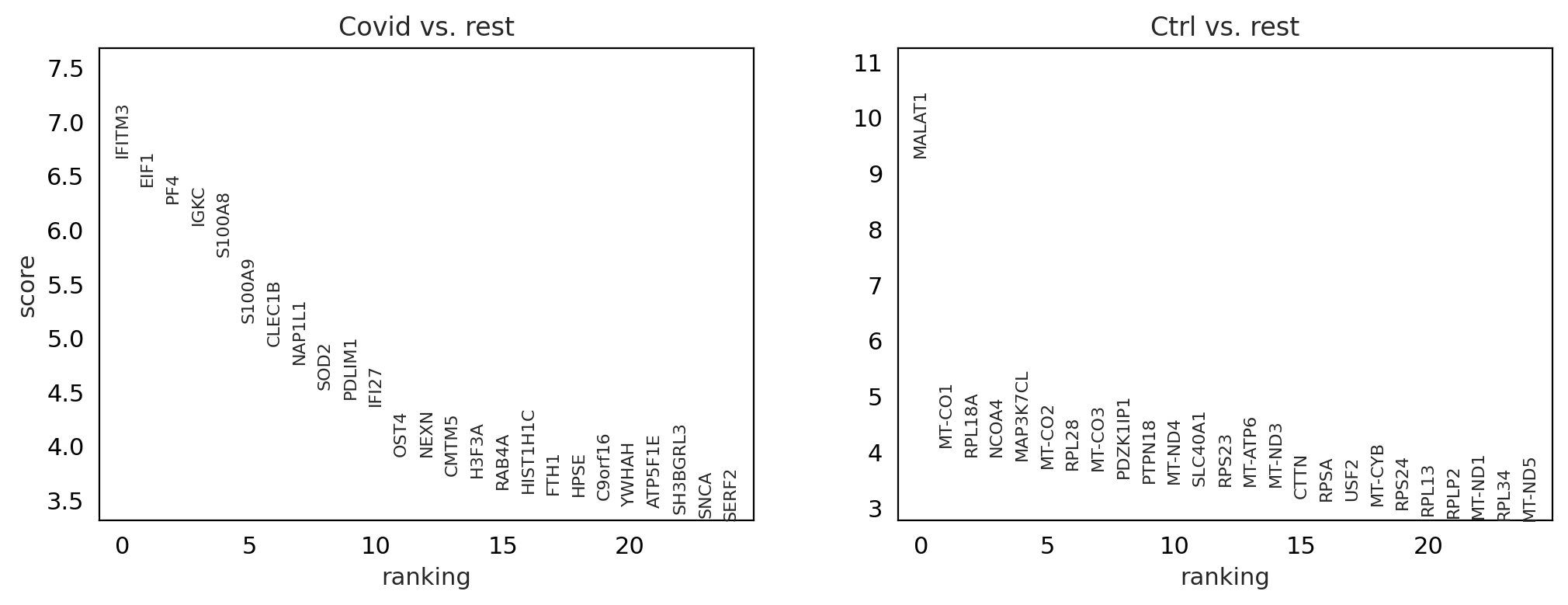
sc.tl.rank_genes_groups(cl1, 'type', method='wilcoxon', key_added = "wilcoxon")
sc.pl.rank_genes_groups(cl1, n_genes=25, sharey=False, key="wilcoxon")
ranking genes
finished: added to `.uns['wilcoxon']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:02)
#@title save differetial expressed data
save_file = 'Objects/sc_QCNFSDM_scCorrected__clustered_DGE_covid.h5ad'
adata.write_h5ad(save_file)