
Pre-processing#
Quality Control#
Remove Empty Droplet using drokick#
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch'
var: 'gene_ids', 'feature_types', 'genome'
#@title Dropkick run
# To skip running this code cell run the following command
%%script echo skipping
# simple preprocessing of anndata object to get metrics for plot
adata = dk.recipe_dropkick(adata, n_hvgs=None, X_final="raw_counts")
skipping
print(adata)
print("===================================================\n")
# print(adata_empty)
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch'
var: 'gene_ids', 'feature_types', 'genome'
===================================================
%%script echo skipping
qc_plt = dk.qc_summary(adata)
skipping
We can see from the total counts profile (top) that this sample has relatively low background (“empty” droplets), as indicated by the steep dropoff in counts and genes around barcode 5,000.
The dropout rate distribution also looks pretty good, with a steep increase in dropout rate following the first few highly-expressed genes (MALAT1, RPS27, B2M)
This tool preprocesses the data to automatically calculate thresholds on global metrics, then trains a glmnet-style logistic regression model to determine scores and labels for each barcode in the dataset describing likelihood of being a real cell (rather than empty droplet). The dropkick model is returned (adata_model), and contains scikit-learn-style attributes for further exploration. Scores and metrics are also added to the original AnnData object as part of the dk.dropkick function.
The n_jobs parameter will parallelize the model training. Since the default is 5-fold cross-validation, we’ll use 5 cores to quickly train dropkick.
%%script echo skipping
%time adata_model = dk.dropkick(adata, n_jobs=1)
skipping
Running dropkick interactively adds the following attributes to the input AnnData object:
.obs[‘dropkick_score’] Probability of being a real cell Values closer to 0 indicate empty droplets with high ambient RNA content
.obs[‘dropkick_label’] Binary label generated by using cutoff of 0.5 on ‘dropkick_score’ Dropkick’s best estimate of a “good” dataset User is encouraged to adjust the cutoff according to whether they are more concerned with false negatives or false positives
.obs[
] Metrics used for thresholding (metrics parameter, default [‘arcsinh_n_genes_by_counts’], are kept
%%script echo skipping
print(adata.obs['dropkick_score'])
print("=================================================")
print(adata.obs['dropkick_label'])
skipping
The score_plot function displays these barcodes in a scatter plot of ambient percentage (as calculated in the qc_summary plot above) versus total genes detected for each cell. The automated thresholds on the latter heuristic are overlayed to show the initial training labels given to the dropkick model.
%%script echo skipping
score_plt = dk.score_plot(adata)
skipping
%%script echo skipping
dk.coef_inventory(adata)
skipping
%%script echo skipping
coef_plt = dk.coef_plot(adata)
skipping
%%script echo skipping
print("Before empty drop removal :", adata.shape)
print("======================================================")
# make a copy of our AnnData object as adata_nonempty , keeping all barcodes kept by dropkick
adata = adata[(adata.obs.dropkick_label=="True"), :].copy()
print("After empty drop removal :", adata.shape)
skipping
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch'
var: 'gene_ids', 'feature_types', 'genome'
Calculate QC#
Steps :
Filter Zero count cells / cell with low number of detected genes
Correction of Ambient RNA
Doublet Detection
Normalization (Pearson Residual transformation)
Feature selection
Dimensionality reduction
Note : In order to delete low quality cells , we have to jointly considered following qc metrices (so we don’t remove much cells) :
Count Depth (if low means small or dying cell)
Few genes per cell (dying cell)
High mitochondrial genes fraction per cell (Mitochondria leaked
out and cell ruptured)
Solution : using MAD (median absolute deviations) automatic method
Mark cell oulier > MAD >= 5 threshold
#@title Calculate QC Metrics
# First Calculate QC Metrices for [ mt, ribo, hb ] and Counts related metrices
###==================================###########
# We calculate QC metrices for Mitochondrial fraction , ribosomal fraction and haemoglobin fraction
# And we have count related metrcies like Depth count (Total count) , n_genes_by_counts , pct_counts_mt
# We use MAD (Median Deviation) of these metrices with threshold like nmads = 3 and represent cells as outlier
# Then we use these ouliers cells to remove So we achieve filtering
###==================================###########
# mitochondrial genes
adata.var["mt"] = adata.var_names.str.startswith("MT-")
# ribosomal genes
adata.var["ribo"] = adata.var_names.str.startswith(("RPS", "RPL"))
# hemoglobin genes.
adata.var["hb"] = adata.var_names.str.contains(("^HB[^(P)]"))
adata.var.head()
| gene_ids | feature_types | genome | mt | ribo | hb | |
|---|---|---|---|---|---|---|
| MIR1302-2HG | ENSG00000243485 | Gene Expression | GRCh38 | False | False | False |
| FAM138A | ENSG00000237613 | Gene Expression | GRCh38 | False | False | False |
| OR4F5 | ENSG00000186092 | Gene Expression | GRCh38 | False | False | False |
| AL627309.1 | ENSG00000238009 | Gene Expression | GRCh38 | False | False | False |
| AL627309.3 | ENSG00000239945 | Gene Expression | GRCh38 | False | False | False |
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb'
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt", "ribo", "hb"], inplace=True, percent_top=[20], log1p=False
)
adata
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch', 'n_genes_by_counts', 'total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'pct_counts_hb'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
n_genes_by_counts in .obs is the number of genes with positive counts in a cell,
total_counts is the total number of counts for a cell, this might also be known as library size, and
pct_counts_mt is the proportion of total counts for a cell which are mitochondrial.
adata.obs.head(3)
| type | sample | batch | n_genes_by_counts | total_counts | pct_counts_in_top_20_genes | total_counts_mt | pct_counts_mt | total_counts_ribo | pct_counts_ribo | total_counts_hb | pct_counts_hb | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | Covid | covid_1 | 0 | 2140 | 7698.0 | 24.123149 | 525.0 | 6.819953 | 2564.0 | 33.307353 | 2.0 | 0.025981 |
| TACCCACAGCGGGTTA-1-0 | Covid | covid_1 | 0 | 3391 | 13416.0 | 21.414729 | 952.0 | 7.096005 | 2264.0 | 16.875373 | 6.0 | 0.044723 |
| CCCAACTTCATATGGC-1-0 | Covid | covid_1 | 0 | 3654 | 16498.0 | 22.402715 | 1253.0 | 7.594860 | 2723.0 | 16.505031 | 1.0 | 0.006061 |
Plot QC#
Now we can Plot these calculated metrices . each dot represents cell in plot . Group by sample , so we can see for each sample how merics behaves
sc.pl.violin(adata, ['total_counts', 'n_genes_by_counts', 'pct_counts_mt','pct_counts_ribo', 'pct_counts_hb'],
jitter=0.4, groupby = 'sample', rotation= 45)
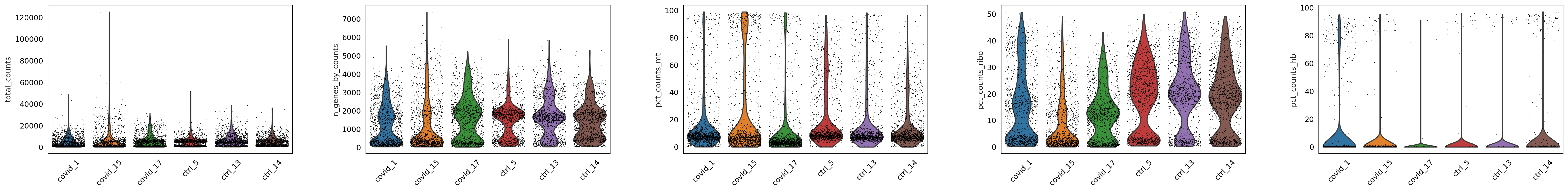
As you can see, there is quite some difference in quality for the 4 datasets, with for instance the covid_15 sample having fewer cells with many detected genes and more mitochondrial content. As the ribosomal proteins are highly expressed they will make up a larger proportion of the transcriptional landscape when fewer of the lowly expressed genes are detected. And we can plot the different QC-measures as scatter plots.
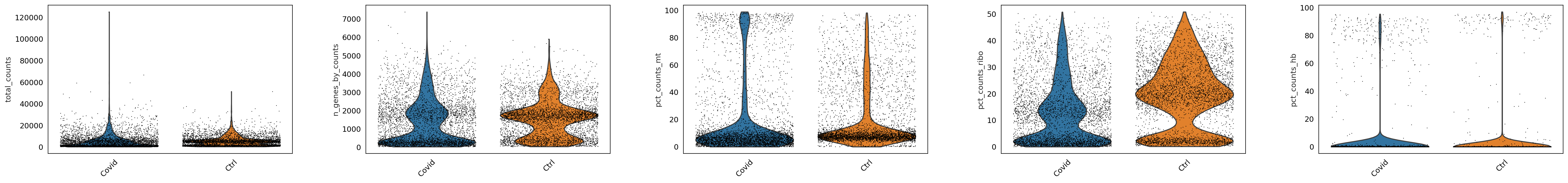
sc.pl.violin(adata, ['total_counts', 'n_genes_by_counts', 'pct_counts_mt','pct_counts_ribo', 'pct_counts_hb'],
jitter=0.4, groupby = 'type', rotation= 45)
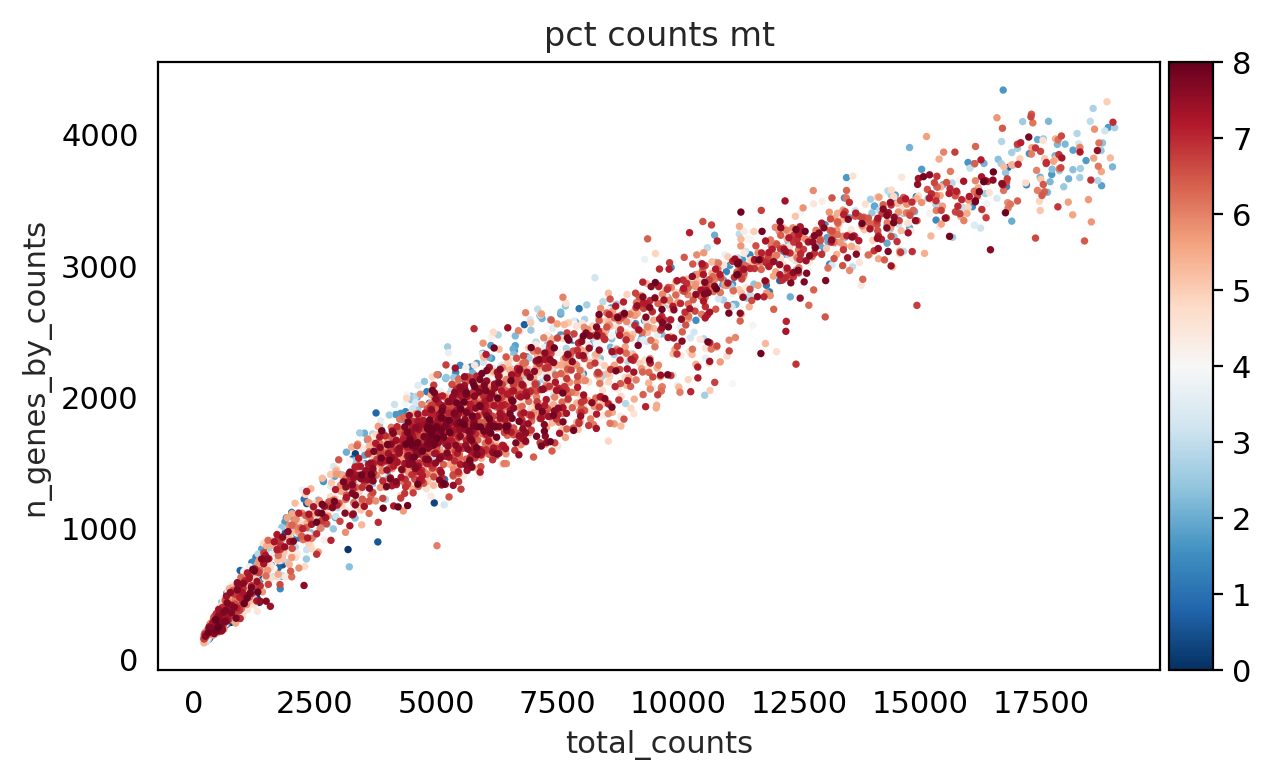
sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt", show= False, legend_loc= "best")
<Axes: title={'center': 'pct counts mt'}, xlabel='total_counts', ylabel='n_genes_by_counts'>
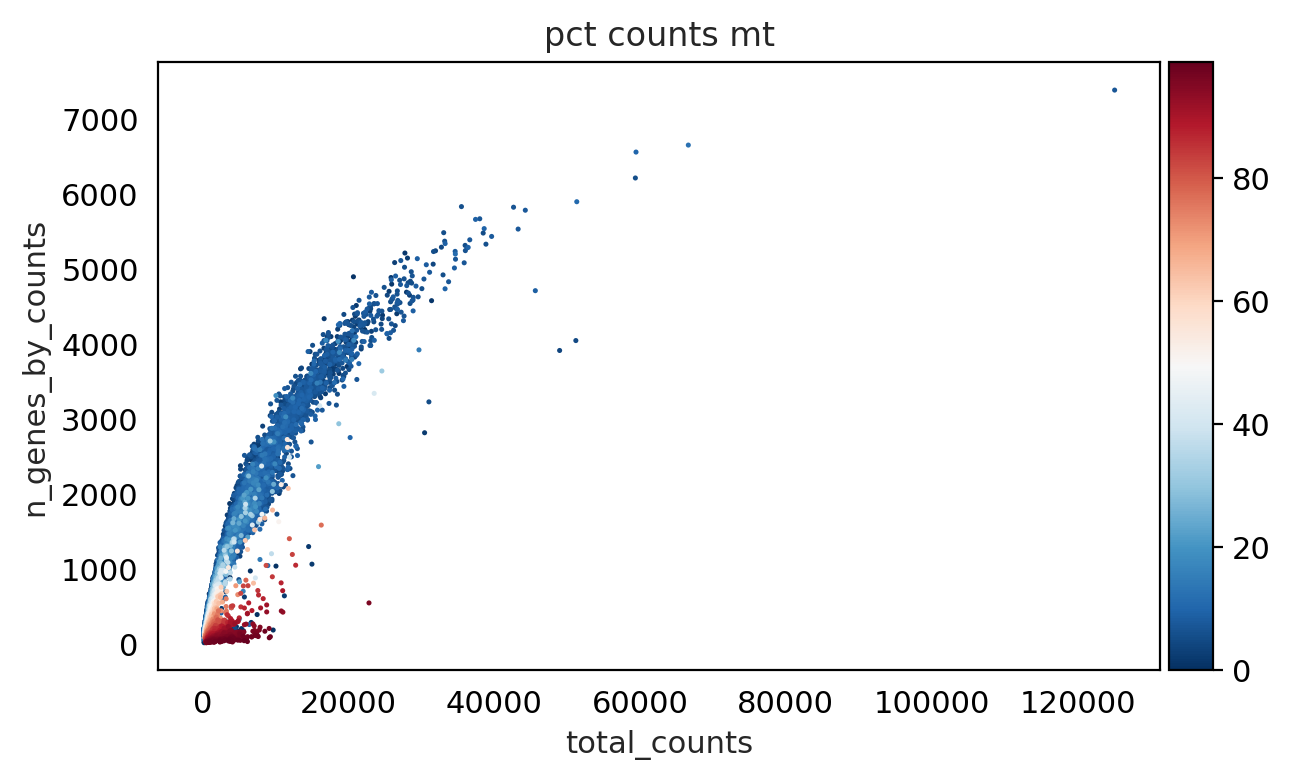
As we can see from above plot : genes with low counts have more percentage mitochondria and some cells have less mitochondria .
Filtering (Joint Effect)#
We have to filter cells by considering joint effects by these WC metrics we have calculated in previous cells .
We use Automatic method MAD : which depends on Median and Standard deviation
We first calculate outliers by each od these QC effects
Filter Using Automatic Method : MAD#
#@title Create outlier function
# function that takes a metric, i.e. a column in .obs and the number of MADs (nmad)
# Function take 3 Arguments : 1) Adata object , 2) Metric colum for condition 3) nmads = 3 or 5 or ...?
def is_outlier(adata, metric: str, nmads: int):
M = adata.obs[metric]
outlier = (M < np.median(M) - nmads * median_abs_deviation(M)) | (
np.median(M) + nmads * median_abs_deviation(M) < M
)
return outlier
adata.obs.head(3)
| type | sample | batch | n_genes_by_counts | total_counts | pct_counts_in_top_20_genes | total_counts_mt | pct_counts_mt | total_counts_ribo | pct_counts_ribo | total_counts_hb | pct_counts_hb | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | Covid | covid_1 | 0 | 2140 | 7698.0 | 24.123149 | 525.0 | 6.819953 | 2564.0 | 33.307353 | 2.0 | 0.025981 |
| TACCCACAGCGGGTTA-1-0 | Covid | covid_1 | 0 | 3391 | 13416.0 | 21.414729 | 952.0 | 7.096005 | 2264.0 | 16.875373 | 6.0 | 0.044723 |
| CCCAACTTCATATGGC-1-0 | Covid | covid_1 | 0 | 3654 | 16498.0 | 22.402715 | 1253.0 | 7.594860 | 2723.0 | 16.505031 | 1.0 | 0.006061 |
#@title use QC metrices to find outlier cells
adata.obs["outlier"] = (
is_outlier(adata, "total_counts", 5)
| is_outlier(adata, "n_genes_by_counts", 5)
| is_outlier(adata, "pct_counts_in_top_20_genes", 5)
)
print(adata.obs.outlier.value_counts())
adata.obs.head(3)
False 6898
True 2102
Name: outlier, dtype: int64
| type | sample | batch | n_genes_by_counts | total_counts | pct_counts_in_top_20_genes | total_counts_mt | pct_counts_mt | total_counts_ribo | pct_counts_ribo | total_counts_hb | pct_counts_hb | outlier | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | Covid | covid_1 | 0 | 2140 | 7698.0 | 24.123149 | 525.0 | 6.819953 | 2564.0 | 33.307353 | 2.0 | 0.025981 | False |
| TACCCACAGCGGGTTA-1-0 | Covid | covid_1 | 0 | 3391 | 13416.0 | 21.414729 | 952.0 | 7.096005 | 2264.0 | 16.875373 | 6.0 | 0.044723 | False |
| CCCAACTTCATATGGC-1-0 | Covid | covid_1 | 0 | 3654 | 16498.0 | 22.402715 | 1253.0 | 7.594860 | 2723.0 | 16.505031 | 1.0 | 0.006061 | False |
#@title use Mitochondrial QC metric to find outlier cells
adata.obs["mt_outlier"] = is_outlier(adata, "pct_counts_mt", 3) | (
adata.obs["pct_counts_mt"] > 8 #cells with a percentage of mitochondrial counts exceeding 8 % are filtered out
)
print(adata.obs.mt_outlier.value_counts())
adata.obs.head(3)
False 4762
True 4238
Name: mt_outlier, dtype: int64
| type | sample | batch | n_genes_by_counts | total_counts | pct_counts_in_top_20_genes | total_counts_mt | pct_counts_mt | total_counts_ribo | pct_counts_ribo | total_counts_hb | pct_counts_hb | outlier | mt_outlier | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | Covid | covid_1 | 0 | 2140 | 7698.0 | 24.123149 | 525.0 | 6.819953 | 2564.0 | 33.307353 | 2.0 | 0.025981 | False | False |
| TACCCACAGCGGGTTA-1-0 | Covid | covid_1 | 0 | 3391 | 13416.0 | 21.414729 | 952.0 | 7.096005 | 2264.0 | 16.875373 | 6.0 | 0.044723 | False | False |
| CCCAACTTCATATGGC-1-0 | Covid | covid_1 | 0 | 3654 | 16498.0 | 22.402715 | 1253.0 | 7.594860 | 2723.0 | 16.505031 | 1.0 | 0.006061 | False | False |
#@title Filter out cells with Above QC metrices :
print(f"Total number of cells Before Filtering : {adata.n_obs}")
adata = adata[(~adata.obs.outlier) & (~adata.obs.mt_outlier)].copy()
print("")
print(f"Number of cells after filtering of low quality cells: {adata.n_obs}")
Total number of cells Before Filtering : 9000
Number of cells after filtering of low quality cells: 4205
Plot Filtered QC#
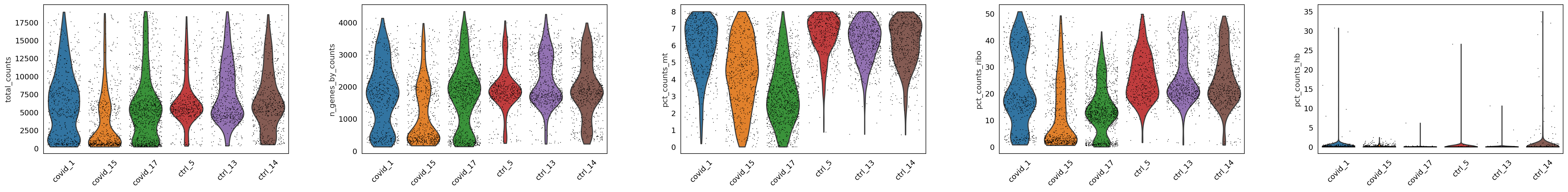
#@title Filtered Data Plot
sc.pl.violin(adata, ['total_counts', 'n_genes_by_counts', 'pct_counts_mt','pct_counts_ribo', 'pct_counts_hb'],
jitter=0.4, groupby = 'sample', rotation= 45)
sc.pl.scatter(adata, "total_counts", "n_genes_by_counts", color="pct_counts_mt", show= False, legend_loc= "best")
<Axes: title={'center': 'pct counts mt'}, xlabel='total_counts', ylabel='n_genes_by_counts'>
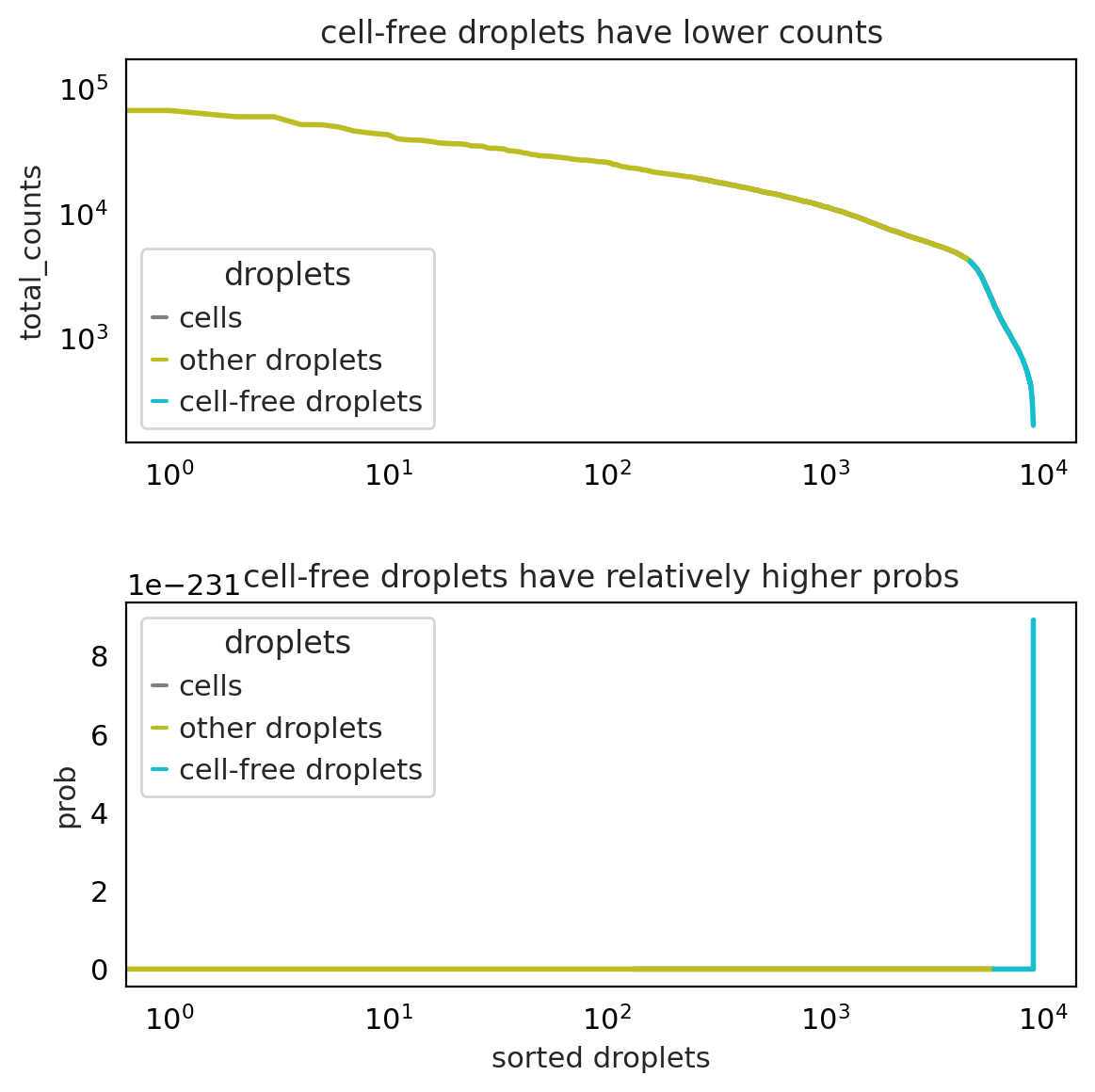
ambient RNA correction using scAR :#
To work with scAR we need following :
raw Anndata (unfitered from sequencing machine , which contain Empty droplets and cells with only free mRNA )
Filtered data
Then we estimate Ambient profile from raw and filtered data
Then we use Machine learning to train data to identify Ambient RNA and then we correct count matrix with respect to Ambient profile
setup_anndata is inspired by EmptyDrops which assumes that cell-free droplets are sampled from a multinomial distribution.
It first calculates an initial ambient profile by averaging all droplets in raw_adata
It tested whether droplets fit the multinomial distribution (the ambient profile as the prob parameter). The relatively high probabilities suggest cell-free droplets
It re-calculates the ambient profile using the identified cell-free droplets
It repeats step 2 and step 3 for iterations
The final ambient profile is saved in adata.uns
#@title Read adata_raw
adata_raw = sc.read_h5ad("/content/drive/MyDrive/scRNA_using_Python/Objects/adata_raw_covid.h5ad")
adata_raw
AnnData object with n_obs × n_vars = 9000 × 33538
obs: 'type', 'sample', 'batch'
var: 'gene_ids', 'feature_types', 'genome'
#@title Estimate the ambient profile
setup_anndata(
adata = adata,
raw_adata = adata_raw,
prob = 0.895,
kneeplot = True
)
2023-05-28 05:45:56|INFO|setup_anndata|Randomly sample 9000 droplets from 9000 droplets.
INFO:setup_anndata:Randomly sample 9000 droplets from 9000 droplets.
2023-05-28 05:45:56|INFO|setup_anndata|Estimating ambient profile for ['Gene Expression']
Categories (1, object): ['Gene Expression']...
INFO:setup_anndata:Estimating ambient profile for ['Gene Expression']
Categories (1, object): ['Gene Expression']...
2023-05-28 05:46:04|INFO|setup_anndata|Iteration: 1
INFO:setup_anndata:Iteration: 1
2023-05-28 05:46:12|INFO|setup_anndata|Iteration: 2
INFO:setup_anndata:Iteration: 2
2023-05-28 05:46:20|INFO|setup_anndata|Iteration: 3
INFO:setup_anndata:Iteration: 3
2023-05-28 05:46:20|INFO|setup_anndata|Estimated ambient profile for Gene Expression saved in adata.uns
INFO:setup_anndata:Estimated ambient profile for Gene Expression saved in adata.uns
2023-05-28 05:46:20|INFO|setup_anndata|Estimated ambient profile for all features saved in adata.uns
INFO:setup_anndata:Estimated ambient profile for all features saved in adata.uns
#@title Traine Ambient Data
adata_scar = model(raw_count=adata, # In the case of Anndata object, scar will automatically use the estimated ambient_profile present in adata.uns.
ambient_profile=adata.uns['ambient_profile_Gene Expression'],
feature_type='mRNA',
sparsity=1,
device='cuda' # Both cpu and cuda are supported.
)
adata_scar.train(epochs=200,
batch_size=64,
verbose=True
)
# After training, we can infer the native true signal
adata_scar.inference(batch_size=256) # by defaut, batch_size = None, set a batch_size if getting a memory issue
2023-05-28 05:46:42|INFO|model|cuda will be used.
INFO:model:cuda will be used.
2023-05-28 05:46:46|INFO|VAE|Running VAE using the following param set:
INFO:VAE:Running VAE using the following param set:
2023-05-28 05:46:46|INFO|VAE|...denoised count type: mRNA
INFO:VAE:...denoised count type: mRNA
2023-05-28 05:46:46|INFO|VAE|...count model: binomial
INFO:VAE:...count model: binomial
2023-05-28 05:46:46|INFO|VAE|...num_input_feature: 33538
INFO:VAE:...num_input_feature: 33538
2023-05-28 05:46:46|INFO|VAE|...NN_layer1: 150
INFO:VAE:...NN_layer1: 150
2023-05-28 05:46:46|INFO|VAE|...NN_layer2: 100
INFO:VAE:...NN_layer2: 100
2023-05-28 05:46:46|INFO|VAE|...latent_space: 15
INFO:VAE:...latent_space: 15
2023-05-28 05:46:46|INFO|VAE|...dropout_prob: 0.00
INFO:VAE:...dropout_prob: 0.00
2023-05-28 05:46:46|INFO|VAE|...expected data sparsity: 1.00
INFO:VAE:...expected data sparsity: 1.00
2023-05-28 05:46:46|INFO|model|kld_weight: 1.00e-05
INFO:model:kld_weight: 1.00e-05
2023-05-28 05:46:46|INFO|model|learning rate: 1.00e-03
INFO:model:learning rate: 1.00e-03
2023-05-28 05:46:46|INFO|model|lr_step_size: 5
INFO:model:lr_step_size: 5
2023-05-28 05:46:46|INFO|model|lr_gamma: 0.97
INFO:model:lr_gamma: 0.97
Training: 100%|██████████| 200/200 [03:38<00:00, 1.09s/it, Loss=5.4521e+03]
#@title Denoised counts :
denoised_count = pd.DataFrame(adata_scar.native_counts, index=adata.obs_names, columns=adata.var_names)
print("")
print(denoised_count.shape)
print("=================================================")
denoised_count.head()
(4205, 33538)
=================================================
| MIR1302-2HG | FAM138A | OR4F5 | AL627309.1 | AL627309.3 | AL627309.2 | AL627309.4 | AL732372.1 | OR4F29 | AC114498.1 | ... | AC007325.2 | BX072566.1 | AL354822.1 | AC023491.2 | AC004556.1 | AC233755.2 | AC233755.1 | AC240274.1 | AC213203.1 | FAM231C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| TACCCACAGCGGGTTA-1-0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CCCAACTTCATATGGC-1-0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ATTCCTAGTGACTGTT-1-0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CCTAAGACAGATTAAG-1-0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
5 rows × 33538 columns
#@title Add Denoised count (Ambient RNA corrected ) to adata :
adata
adata.layers["counts"] = adata.X
adata.layers["ambient_counts"] = adata_scar.native_counts
adata.X = adata.layers["ambient_counts"]
adata
AnnData object with n_obs × n_vars = 4205 × 33538
obs: 'type', 'sample', 'batch', 'n_genes_by_counts', 'total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'pct_counts_hb', 'outlier', 'mt_outlier'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
uns: 'sample_colors', 'type_colors', 'ambient_profile_Gene Expression', 'ambient_profile_all'
layers: 'counts', 'ambient_counts'
adata.var.head()
| gene_ids | feature_types | genome | mt | ribo | hb | n_cells_by_counts | mean_counts | pct_dropout_by_counts | total_counts | |
|---|---|---|---|---|---|---|---|---|---|---|
| MIR1302-2HG | ENSG00000243485 | Gene Expression | GRCh38 | False | False | False | 0 | 0.000000 | 100.000000 | 0.0 |
| FAM138A | ENSG00000237613 | Gene Expression | GRCh38 | False | False | False | 0 | 0.000000 | 100.000000 | 0.0 |
| OR4F5 | ENSG00000186092 | Gene Expression | GRCh38 | False | False | False | 0 | 0.000000 | 100.000000 | 0.0 |
| AL627309.1 | ENSG00000238009 | Gene Expression | GRCh38 | False | False | False | 25 | 0.002778 | 99.722222 | 25.0 |
| AL627309.3 | ENSG00000239945 | Gene Expression | GRCh38 | False | False | False | 1 | 0.000111 | 99.988889 | 1.0 |
Filter Genes#
#@title Filter genes :
print(f"Total number of genes: {adata.n_vars}")
# Min 20 cells - filters out 0 count genes
sc.pp.filter_genes(adata, min_cells=20)
print(f"Number of genes after cell filter: {adata.n_vars}")
Total number of genes: 33538
Number of genes after cell filter: 7662
# As we have . we can remove genes correspond to Mirochondria , Ribosomal and MALAT1
# Because High level of MALAT1 and MT referes to Technical variation
# group genes with MALAT1 , MT and HB and remove
malat1 = adata.var_names.str.startswith('MALAT1')
mito_genes = adata.var_names.str.startswith('MT-')
hb_genes = adata.var_names.str.contains('^HB[^(P)]')
remove = np.add(mito_genes, malat1)
remove = np.add(remove, hb_genes)
keep = np.invert(remove)
print("before removal of genes :" , adata.n_obs, adata.n_vars)
adata = adata[:,keep]
print("After removal of genes :" ,adata.n_obs, adata.n_vars)
before removal of genes : 4205 7662
After removal of genes : 4205 7650
#@title install pybiomart
Cell Cycle Scoring#
#@title Cell Cycle scoring :
!if [ ! -f data/regev_lab_cell_cycle_genes.txt ]; then curl -o data/regev_lab_cell_cycle_genes.txt https://raw.githubusercontent.com/theislab/scanpy_usage/master/180209_cell_cycle/data/regev_lab_cell_cycle_genes.txt; fi
cell_cycle_genes = [x.strip() for x in open('./data/regev_lab_cell_cycle_genes.txt')]
print(len(cell_cycle_genes))
# Split into 2 lists
s_genes = cell_cycle_genes[:43]
g2m_genes = cell_cycle_genes[43:]
cell_cycle_genes = [x for x in cell_cycle_genes if x in adata.var_names]
print(len(cell_cycle_genes))
97
33
# save normalized counts in raw slot.
adata.cycle = adata
# normalize to depth 10 000
sc.pp.normalize_per_cell(adata.cycle, counts_per_cell_after=1e4)
# logaritmize
sc.pp.log1p(adata.cycle)
# scale
sc.pp.scale(adata.cycle)
sc.tl.score_genes_cell_cycle(adata.cycle, s_genes=s_genes, g2m_genes=g2m_genes)
adata.cycle.obs.phase.value_counts()
G1 1474
G2M 1371
S 1360
Name: phase, dtype: int64
sc.pl.violin(adata.cycle, ['S_score', 'G2M_score'],
jitter=0.4, rotation=45)
Doublet detection#
#@title Doublet Detection :
print(adata.shape)
(4205, 7650)
# Change the layer of count to Ambient counts
adata.X = adata.layers["ambient_counts"]
#@title Run scvi setup model
scvi.model.SCVI.setup_anndata(adata)
vae = scvi.model.SCVI(adata)
vae.train()
INFO:pytorch_lightning.utilities.rank_zero:GPU available: True (cuda), used: True
INFO:pytorch_lightning.utilities.rank_zero:TPU available: False, using: 0 TPU cores
INFO:pytorch_lightning.utilities.rank_zero:IPU available: False, using: 0 IPUs
INFO:pytorch_lightning.utilities.rank_zero:HPU available: False, using: 0 HPUs
INFO:pytorch_lightning.accelerators.cuda:LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 400/400: 100%|██████████| 400/400 [02:27<00:00, 2.70it/s, loss=3.52e+03, v_num=1]
INFO:pytorch_lightning.utilities.rank_zero:`Trainer.fit` stopped: `max_epochs=400` reached.
Epoch 400/400: 100%|██████████| 400/400 [02:27<00:00, 2.71it/s, loss=3.52e+03, v_num=1]
#@title Train solo model
solo = scvi.external.SOLO.from_scvi_model(vae)
solo.train()
INFO Creating doublets, preparing SOLO model.
INFO:pytorch_lightning.utilities.rank_zero:GPU available: True (cuda), used: True
INFO:pytorch_lightning.utilities.rank_zero:TPU available: False, using: 0 TPU cores
INFO:pytorch_lightning.utilities.rank_zero:IPU available: False, using: 0 IPUs
INFO:pytorch_lightning.utilities.rank_zero:HPU available: False, using: 0 HPUs
INFO:pytorch_lightning.accelerators.cuda:LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 400/400: 100%|██████████| 400/400 [02:03<00:00, 3.24it/s, loss=0.339, v_num=1]
INFO:pytorch_lightning.utilities.rank_zero:`Trainer.fit` stopped: `max_epochs=400` reached.
Epoch 400/400: 100%|██████████| 400/400 [02:03<00:00, 3.23it/s, loss=0.339, v_num=1]
#@title Predict the Doublet
df = solo.predict()
df['prediction'] = solo.predict(soft = False)
df.index = df.index.map(lambda x: x[:-2])
df
| doublet | singlet | prediction | |
|---|---|---|---|
| AGGGTCCCATGACCCG-1-0 | -1.240547 | 1.317724 | singlet |
| TACCCACAGCGGGTTA-1-0 | 0.230032 | -0.015135 | doublet |
| CCCAACTTCATATGGC-1-0 | 0.087104 | 0.086796 | doublet |
| ATTCCTAGTGACTGTT-1-0 | -1.723657 | 1.353531 | singlet |
| CCTAAGACAGATTAAG-1-0 | -1.451537 | 1.859437 | singlet |
| ... | ... | ... | ... |
| GCTGCAGTCTGTCAGA-14-5 | -1.007731 | 0.915832 | singlet |
| GAGGCCTTCTCCTGCA-14-5 | -0.019297 | 0.247610 | singlet |
| CCCTAACAGTTTCTTC-14-5 | -0.336324 | -0.266451 | singlet |
| GGGATGATCAAGCTTG-14-5 | 0.087399 | -0.348760 | doublet |
| CAATGACCACTGCATA-14-5 | 0.385331 | -0.194242 | doublet |
4205 rows × 3 columns
#@title Number of Doublets:
len(df[df.prediction == 'doublet']) # pretty high number
669
#THIS STEP IS NOT NECESSARY, just to visuallize results
adata_doublet = adata
adata_doublet
AnnData object with n_obs × n_vars = 4205 × 7650
obs: 'type', 'sample', 'batch', 'n_genes_by_counts', 'total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'pct_counts_hb', 'outlier', 'mt_outlier', 'n_counts', 'S_score', 'G2M_score', 'phase', '_scvi_batch', '_scvi_labels'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'mean', 'std'
uns: 'sample_colors', 'type_colors', 'ambient_profile_Gene Expression', 'ambient_profile_all', 'log1p', '_scvi_uuid', '_scvi_manager_uuid'
layers: 'counts', 'ambient_counts'
adata_doublet.obs['prediction'] = df.prediction
sc.pp.normalize_total(adata_doublet, target_sum = 1e4)
sc.pp.log1p(adata_doublet)
sc.tl.pca(adata_doublet)
sc.pp.neighbors(adata_doublet)
sc.tl.umap(adata_doublet)
sc.tl.leiden(adata_doublet, resolution = 0.5)
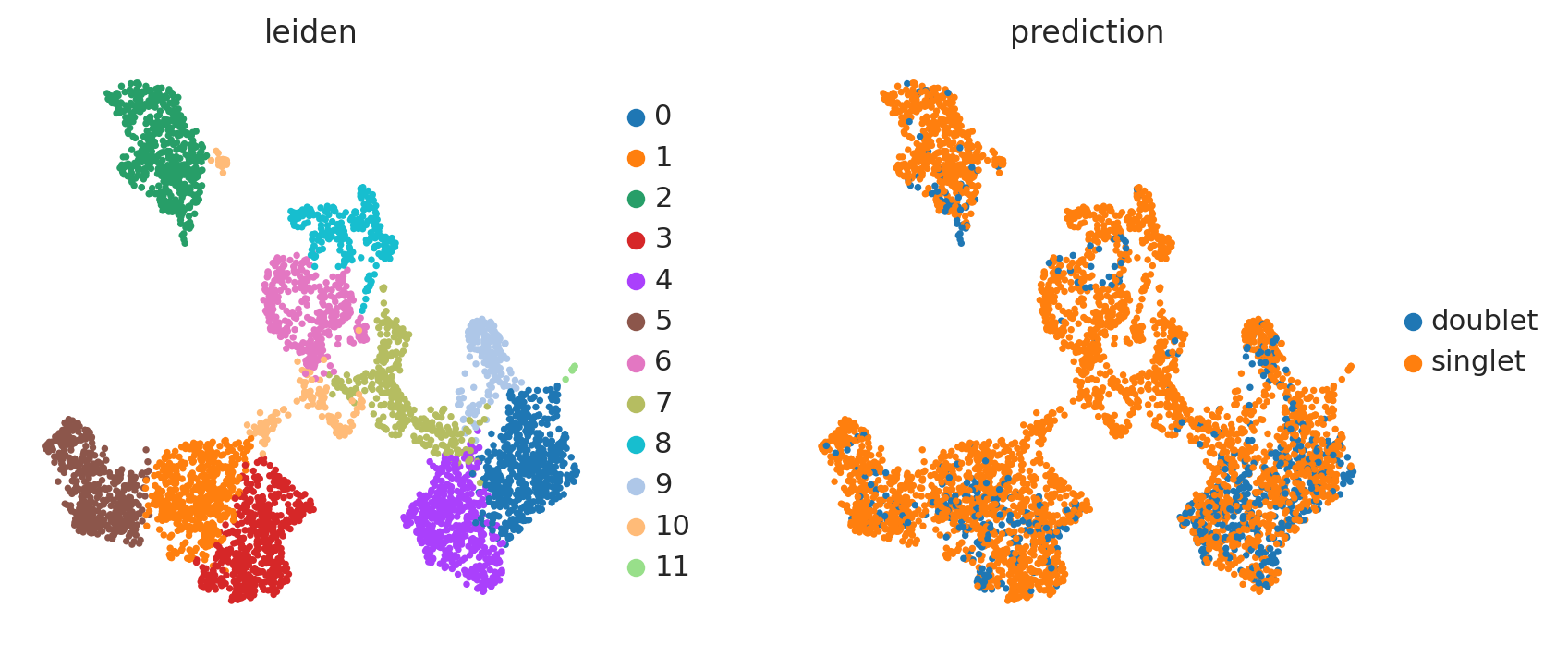
with rc_context({'figure.figsize': (4, 4)}):
sc.pl.umap(adata_doublet, color = ['leiden', 'prediction'])
doublet_d = dict(zip(df.index, df.prediction))
def doublet_function(x):
try:
return doublet_d[x]
except:
return 'filtered'
adata.obs['doublet'] = adata.obs.index.map(doublet_function)
adata
AnnData object with n_obs × n_vars = 4205 × 7650
obs: 'type', 'sample', 'batch', 'n_genes_by_counts', 'total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'pct_counts_hb', 'outlier', 'mt_outlier', 'n_counts', 'S_score', 'G2M_score', 'phase', '_scvi_batch', '_scvi_labels', 'prediction', 'leiden', 'doublet'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'mean', 'std'
uns: 'sample_colors', 'type_colors', 'ambient_profile_Gene Expression', 'ambient_profile_all', 'log1p', '_scvi_uuid', '_scvi_manager_uuid', 'pca', 'neighbors', 'umap', 'leiden', 'leiden_colors', 'prediction_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'ambient_counts'
obsp: 'distances', 'connectivities'
#@title remove predicted doublets
adata = adata[adata.obs.doublet == 'singlet']
adata
View of AnnData object with n_obs × n_vars = 3536 × 7650
obs: 'type', 'sample', 'batch', 'n_genes_by_counts', 'total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'pct_counts_ribo', 'total_counts_hb', 'pct_counts_hb', 'outlier', 'mt_outlier', 'n_counts', 'S_score', 'G2M_score', 'phase', '_scvi_batch', '_scvi_labels', 'prediction', 'leiden', 'doublet'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'ribo', 'hb', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'mean', 'std'
uns: 'sample_colors', 'type_colors', 'ambient_profile_Gene Expression', 'ambient_profile_all', 'log1p', '_scvi_uuid', '_scvi_manager_uuid', 'pca', 'neighbors', 'umap', 'leiden', 'leiden_colors', 'prediction_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'ambient_counts'
obsp: 'distances', 'connectivities'
Save QC Filtered Data#
save_file = 'Objects/sc_qc_filtered_covid.h5ad'
adata.write_h5ad(save_file)